


Autoregressive Biomedicine
Reimagining Life Science Through Next-Token Prediction

Introduction
Biology studies complex and hierarchically organized systems: Biomolecules, cells, tissues, organs, organisms, and ecosystems. Cataloging these systems, their constituents, and behaviors is a foundational component of research, harkening back to the early days of Linnaean scientific classification. Today, we have taxonomies, ontologies, encyclopedias, databases, biobanks, and atlases with an astonishing number of entries.
Data affirms or negates hypotheses, it doesn’t explain itself. Human ingenuity is responsible for interpretation and for formulating the next questions that drive science forward. But our ingenuity has limits. Though a flood of new data advances individual domains, it exacerbates the challenge of maintaining a holistic view. One that spans from molecules to patients and everything between.
At the frontier of AI research, large language models (LLMs) have been trained on vast swathes of the internet’s text and are moving on to images, videos, audio, and 3D spaces. There’s optimism in some corners that scaled up multimodal LLMs are a viable route to the first incarnation of powerful AI1. It’s easy to claim that this development will transform the sciences, including biomedicine. A precise articulation of how is more elusive.
Imagine a hypothetical AI system from the near future, a next-token prediction model trained not only on internet data but also on the full catalog of biomedical data. That is, every DNA sequence, protein structure, single cell omics profile, microscopy and clinical image, patient health record, and more. Given access to this AI, what should we want it to achieve? Can it make sense of the data we’ve accumulated? And how will that impact the practice of biomedicine in the lab and in the clinic?
To answer these questions, I’ll start with a review of how weaker AI models are currently changing biomedical research, covering recent progress in unimodal and multimodal learning. Then, I’ll speculate on how integrative modeling promised by advanced AI might accelerate biomedicine as a data alchemist, world model, and translational reasoner. Finally, I examine prospects and opportunities for realizing this future.
Review
Note: Feel free to skim or skip ahead. I suggest looking through at least the Autoregressive multimodal models (AMMs) subsection before moving on to Capabilities of a powerful biomedical AI. The review adds context and justification for later arguments – and is hopefully interesting – but non-essential.
AI in biomedicine: One modality at a time
AlphaFold22 marked a watershed moment for AI in biomedicine by cracking protein structure prediction. It spurred the pursuit of equally profound breakthroughs for genetics, omics, therapeutics, and clinical imaging. AI foundation models now exist for numerous modalities with applications in basic and clinical research. To navigate this rapidly evolving landscape, let's survey it by working our way up through the levels of biological organization.
DNA, RNA, and proteins
We begin with DNA—life's primary information storage unit and the code for making RNAs and proteins, its functional building blocks. RNAs are involved in protein synthesis, gene regulation, and catalysis, while proteins give a cell structure, catalyze biochemical reactions as enzymes, and transmit signals.
The human genome, with roughly 3 billion base pairs, is well-modeled by long context language models where each nucleotide (A, C, T, or G) is a token. HyenaDNA3 and Evo24 are state-space models that have scaled next-token prediction to sequences longer than 100K bases and organically learn to annotate genes, pseudogenes, RNA types, and regulatory elements. Evo2 also generates coherent whole genomes with realistic protein coding regions and accurately detects harmful genetic variants.
RNA sequences, derived from fragments of DNA, are suitable for regular transformers. RNA-FM5 and RNAErnie6 learn representations that encode function, structure (via RhoFold+7), and RNA-RNA interactions. Despite the importance of DNA and RNA, knowledge of their intrinsic "grammars" can only go so far. Protein sequences, augmented with rich structural and functional data in the Protein Data Bank (PDB), are more fertile ground for impactful applications.
The AI for proteins ecosystem encompasses myriad tasks and methods. The simplest are sequence-based language models trained on amino acid sequences or multiple sequence alignments (i.e., groups of similar genes found in different species aligned to a common reference)8910. Protein language models (PLMs) yield transferable representations that support folding and designing structures, docking small molecules11, and inferring function and sub-cellular localization12. Structures themselves can be decomposed into 1D sequences of tokens based on motif libraries or learned codebooks to train a PLM, with some benefit for mutational effect prediction131415.
Bespoke and task-specific supervised learning architectures directly optimize for an application rather than using intermediate embeddings. They target protein design1617181920, small molecule docking2122, directed evolution23, function prediction24, molecular dynamics simulation25, and all-atom structure prediction262728 2930.
Single cells
Cells are the fundamental units of life, integrating the information of DNA and the functions of RNAs and proteins. Modern single-cell technologies profile millions of individual cells, with precise measurement of molecular features including gene expression (RNA-seq), chromatin accessibility (ATAC-seq), protein abundance (CITE-seq), genetic variations, morphology (Cell Painting), DNA methylation, DNA 3D shape (Hi-C), metabolites, and lipids. Together they offer comprehensive insights into cellular heterogeneity, regulation, development, and disease states – while somewhat bounded by cost and throughput.
Gene expression, or transcriptomics, is the dominant single cell modality. Here, each cell is represented as a high-dimensional vector: Dimensions (~60K) represent genes and values indicate the corresponding count of measured mRNA transcripts. Vectors are sparse, with only a portion of genes being actively transcribed. The activation pattern of genes defines cell type and state.
Foundation models for transcriptomics adapt language modeling architectures and objectives. A key obstacle is converting vectors into sequences that a transformer can parse. One approach treats genes as learnable tokens and orders them by expression level, placing the most active at the start of the sequence3132. Alternatives use biologically-informed tokens such as embeddings of each gene's amino acid sequence or document embeddings from gene text descriptions, thereby subtly incorporating protein-level or pathway-level information into the model3334. Masked and autoregressive language models trained on the tokenized sequences learn how to integrate data across experimental batches, classify cell types, and simulate expression changes in response to perturbations like the knockout or activation of a gene.
Other single cell modalities supply complementary information to transcriptomics, but smaller datasets have hindered the implementation of foundation models.
Notable exceptions have surfaced in several areas. GET uses chromatin accessibility data to infer gene expression patterns in new biological contexts35. For epigenetics, CpGPT applies masked language modeling to DNA methylation data, uncovering biological signals that correlate with aging and disease risk factors36. In cellular imaging, vision transformers trained on fluorescence microscopy images are used to create “phenomics maps”373839. Maps that relate genetic and chemical perturbations through shared visual signatures and highlight common mechanisms of action.
Tissues, organs, and organ systems
Single cell omics have reshaped basic research, but clinical diagnostics still tend to operate at higher levels of organization: tissues, organs, and organ systems.
Tissues, the functional units of organs, are organized communities of closely interacting cells. Microscopy of H&E-stained histopathology slides is the gold standard for tissue analysis. Slides show individual cells, their local context, and the surrounding tissue architecture – patterns that pathologists use to diagnose and grade diseases. Virchow2 trains a self-supervised vision transformer on histopathology images at multiple magnifications40. Linear probes added on the model's embeddings not only match pathologists at tumor grading and cancer subtype identification, but also extract information beyond human perception including treatment responses and patient survival.
Molecular profiling sheds light on tissue function. Bulk tissue sequencing, which measures average gene expression in a population of cells, has been modeled with masked language models to predict gene-gene relationships and clinical outcomes like those derived from histopathology images4142. A downside is that bulk measurements obscure spatial heterogeneity.
Spatial omics technologies bridge the gap by mapping molecular measurements – at nearly cellular resolution (typically 10-100 cells per spot) – to their physical locations in tissue. Unlocking insights into how neighborhoods and physical barriers shape cell behavior and disease progression. Many pathologies originate from abnormal spatial patterns like dysregulated signaling between neighboring cells or destructive local immune responses. Current AI applications to spatial omics focus on two main problems: improving resolution via deconvolution to single-cell granularity4344, and inferring the spatial contexts of dissociated cells4546. Graph-based methods further leverage the spatial structure of the data to cluster tissue regions with similar molecular profiles, potentially corresponding to interesting microenvironments47.
Organs are collections of interacting tissues. Analogous to histopathology, radiology (e.g., X-ray, MRI, and CT) reveals an organ's structure and subunits, though not at cellular resolution. Methods for modeling radiology data are the same as for other image modalities with the occasional added complexity of a third dimension. Models pre-trained with self-supervised learning (SSL) can be tuned to make diagnoses directly or assist radiologists by annotating regions of interest (tumors, malformations, degeneration, etc.)48. Embeddings from these models are predictive of long-term disease risks and an array of biomarkers and general health signatures49.
Time-series measurements of organ function are another diagnostic tool. Electrocardiograms (ECGs), for example, record the heart's electrical activity. ECG-FM uses contrastive learning on these 1D signals to pre-train a transformer that assesses both ECG-apparent conditions and conditions typically requiring blood tests50.
Functional MRI (fMRI) merges temporal and structural imaging by tracking brain activity over time. BrainLM, trained with hybrid masked and autoregressive language modeling, learns through reconstructing fMRI sequences51. Finetuned on UK Biobank data, it infers clinical variables ranging from age to psychiatric conditions (anxiety, neuroticism, PTSD) and highlights distinct functional units in the brain.
Collections of interacting organs form organ systems like the nervous, cardiovascular, and digestive. Readouts from continuous monitoring with wearable or implanted devices have proven to be surprisingly strong indicators of general health. For the cardiovascular system, patient-level SSL embeddings from Apple Watch readings (ECG for heart rhythm and PPG for blood volume) can determine demographics, medication usage patterns, and diagnosed conditions including asthma and depression and even preemptively detect health events like heart failure52. For the endocrine system, an autoregressive model trained on continuous glucose monitoring data shows broad clinical utility outside its primary purpose53. As expected, it excels at diabetes-related predictions. Additionally, it encodes signatures of liver, kidney, and cardiovascular health and holds promise for stratifying clinical trial populations.
An electronic health record (EHR) is the hub for all patient data. It’s an overview of the whole organism. EHRs contain structured data (images, lab values, vital signs) and unstructured text (clinical notes, diagnostic reports). Ambitiously, EHRMamba and DT-GPT try to create “digital twins” by autoregressively generating future health states starting from an EHR5455. They estimate immediate clinical variables and longer-term outcomes including disease progression, hospital readmission, and mortality risk.
The multimodal frontier
Modeling individual modalities has given us some fantastic applications, but it will not help with the big picture. Advances in multimodal AI architectures show ways forward, by learning from the unparalleled variety of data types available in the biomedical domain. This section reviews three architectural paradigms - contrastive learning, multimodal LLMs, and autoregressive multimodal models - and how each has been used to integrate biomedical information and expand the scope of questions we answer with AI.
Contrastive learning
Contrastive learning aligns modalities in the same latent space by pulling paired data points together and pushing non-pairs apart. The canonical example is CLIP, which trains on image-text pairs and projects both modalities into a space where matched images and captions are nearby56. ImageBind57, and related methods5859, extend the framework to image-text, image-video, image-depth, and image-audio with images serving as an anchor modality in all pairs. Despite some modalities never being directly observed together during training (e.g., text and audio), they naturally come into alignment through their relationships with images. Contrastive models excel at zero-shot image classification and cross-modality retrieval tasks.
For biomedical images, BiomedCLIP uses figure-legend pairs from PubMed60. The dataset contains over 30 types of biomedical images including X-rays, MRI, CT scans, and microscopy with descriptive text. Broad training across image types gives superior performance on downstream classification and question-answering tasks compared to specialist models.
The contrastive framework generalizes beyond images. For instance, scCLIP uses gene expression (RNA-seq) and chromatin accessibility (ATAC-seq) data acquired on the same cell as pairs61. While it hasn’t been done yet, the ImageBind idea might be applied to incorporate other readouts commonly paired with gene expression like protein abundance (CITE-seq).
Although it works well for multimodal representation learning, contrastive models cannot generate data or engage in open-ended conversations.
Multimodal LLMs
The exact definition of what constitutes a multimodal LLM is muddled – the definition I use here is a model that processes modalities other than language but only outputs text. Multimodal LLMs join pre-trained language models with modality-specific encoders. They then use either cross-attention or self-attention to fuse multimodal information62636465.
They learn from a combination of paired data and interleaved multimodal documents. Good documents stay focused on a topic and thoughtfully intersperse images and text to convey information. The signal for multimodal fusion comes from the overall context of the document and the proximity of modalities. Both increase the likelihood of finding shared concepts, though the associations may be less obvious than with explicit pairs. Training with documents works better than training solely on paired data, particularly in few-shot settings66.
Cleverly, LLaVa uses language-only LLMs to create synthetic documents that augment a dataset67. It starts by converting an image into a short text description based on some annotations – that could be a string naming objects in the image and their locations. An LLM takes this text and writes a question-answer dialogue about it. For training, the image itself is inserted back into the Q & A document. (I bring this up because I’ll propose a related idea for biomedicine later).
Multimodal LLMs are expert-level analysts of medical images. There are specialist chatbots for histology slides68, CT scans69, and chest X-rays70. LLaVa-Med, in contrast, is a generalist that builds on BiomedCLIP's unified encoder to interpret multiple modalities71. Med-Gemini takes it up a notch by incorporating 3D images, electronic health records, and genetic data in the form of polygenic risk scores (proof-of-concept work also added ECG data)7273.
For molecular data, where descriptive text is scarce, creative techniques for document construction have been devised. CellWhisperer starts with metadata from single cell RNAseq experiments and uses an LLM-based curation pipeline to transform RNA-seq profiles into textual annotations74. As a data analysis assistant, it identifies distinguishing properties of UMAP clusters, classifies cell types, and predicts the demographics (e.g., age) of a cell’s donor. ProteinChatGPT embeds a protein amino acid sequence with a pre-trained PLM and adds it to text tokens75. Trained on protein-question-answer dialogues, it converses about a protein’s function, structure, and taxonomy.
Autoregressive multimodal models
Autoregressive multimodal models (AMMs) reduce everything to next-token prediction. They typically quantize continuous data into discrete tokens and combine them with language tokens in one unified sequence. Essentially, a standard LLM with a multimodal vocabulary. AMMs put multimodality front-and-center from the beginning of training. Unsurprisingly, this enhances multimodal understanding and, remarkably, single modality understanding as well. Uniquely, they learn from data with or without language and generate tokens for any input modality. Architectural simplicity belies impressive capabilities, evidenced by frontier models like Gemini and GPT-4o. The open-source community has only just started to publish and release AMMs7677787980.
Biomedical applications of AMMs are limited but growing. All current implementations focus on modalities readily convertible to text. BioT5, for example, trains on multimodal scientific publications by finding and replacing small molecule and protein references with SMILES strings and FASTA sequences, respectively81. It can be used to generate protein sequences and perform zero- and few-shot property prediction. Tx-LLM also adds nucleic acids and related details about cell types and diseases82. The authors note that this represents an important step toward in silico clinical trials, where there are questions about the intrinsic properties of a therapeutic (e.g., toxicity) as well as its efficacy in context.
In lieu of natural language, gLM2 preserves protein sequences in their native DNA context83. Protein language modeling is then informed by DNA regulatory elements and spatial proximity to other genes – factors that influence expression timing, splicing, and functional relationships. The model is technically multimodal in that it separately tokenizes protein coding regions at the level of amino acids and everything else at the level of nucleotides.
PoET-2 is a multimodal sequence and structure model with autoregressive and masked language modeling decoders84. It’s trained on documents that include a “context” and a “prompt”. For example, the context might be sequences of human antibodies and the prompt a partial sequence. The model then knows to condition its outputs on the context and inpaint the partial sequence to look like a human antibody.
Summary
AI's journey in biomedicine has ushered in modality-specific breakthroughs. Starting with AlphaFold2 for protein structures, AI models have ascended through biological scales – parsing DNA's grammar, deciphering cellular identities, analyzing tissue architecture, interpreting organ function, and constructing patient profiles from EHRs. As unimodal applications mature, three architectural paradigms bridge modalities: contrastive learning frameworks that align paired data in the same latent space; multimodal LLMs that process diverse inputs and generate text; and, AMMs that reduce biological data to tokens within a unified sequence.
Capabilities of a powerful biomedical AI
Putting aside technical and scientific concerns for the sake of curiosity, suppose we have access to a souped up AMM that qualifies as a powerful biomedical AI. The interface is a simple box that takes in text and multimodal data and responses also come out as text and any other task-relevant modalities. Coming back to our original question, how would it revolutionize understanding of biology and medicine?
The envisioned model subsumes the regression, classification, representation learning, and generative tasks mastered by narrow AIs. While such consolidation is valuable, it alone wouldn’t make a profound difference. I propose three modes of intelligence that might: first, a data alchemist that effortlessly translates biological information across modalities; second, a world model capable of constructing digital twins and simulating system dynamics; and third, a translational reasoner that generates hypotheses and navigates causal and multiscale relationships. This is an informed wish list of sorts – speculative and ambitious, but grounded by what AI is achieving in other domains. We will address feasibility in the subsequent Prospects section.
The Data Alchemist
AI transforms information to enhance human interpretation
Discovery is bottlenecked by the difficulty of measurement. Even when armed with the right question, a scientist may spend years tinkering in the lab to find an answer. AI has accelerated the process for certain kinds of questions by inferring the outcome of hard experiments from easier alternatives. AlphaFold, for example, circumvents the multi-year process of crystallizing proteins for cryoEM by folding structures directly from sequence. Crystallography is slow and expensive; sequencing is fast and cheap.
Taking a step back, why does this work? Any two measurements have both mutual and unique information. For proteins, structure is mostly encoded in sequence. However, it isn’t dictated by the sequence alone. Cellular conditions (e.g., chaperones, post-translational modifications) and stochasticity exert influences too. This injects a modicum of uncertainty, but mostly we trust AlphaFold predictions because mutual information remains high for the task.
From a purely information-centric perspective, one might argue that solving protein structures is, and always has been, unnecessary. This view suits AI models, and especially transformers, which are unconcerned with how information is packaged so long as its present. From a human-centric perspective, information is nothing without interpretability: Given a 3D model, a scientist can reason about function or binding sites, not so much with a string of letters. Cognitive biases, which specify the parameters for how and where we perceive patterns, shape what we choose to measure. New data, modalities, and measurement technologies may not strictly provide more information; rather, they offer presentations that align with our mental models of how biology works.
The high-content experimental readouts favored in the life sciences, like omics and images, are perplexing to interpret. Ask a biologist to look at some microscopy images of cells. They may point out distinguishing features: these cells have larger nuclei and those are more circular. Now ask them what the cell being more circular means. Is it dying? Stressed? Getting ready to divide? Is it losing adhesion? Is something degrading, inhibiting, or altering the cytoskeleton? TBD, experiments needed. Circularity is a relatively basic and obvious phenotype too. Other morphological signatures are subtle and unexplained. It would be great to ask an AI to tell us what it knows about the feature in higher resolution, or correlated with additional antibody stains, or even from the perspective of another modality. A data alchemist cannot create information, but it transforms it; extracting maximum insights from a single readout.
A spectrum of mutual information and pairing strength determines the reliability of a transformation
The degree of mutual information and pairing established how faithful and precise a transformation can be. Data pairing runs in a spectrum from strong to weak. At the strong end, data is acquired at the same time and on the same sample, or must be highly reproducible if gathered separately. Examples of strong pairing are multiomics, DNA-RNA, MRI-fMRI, and protein sequence-structure. Weak pairing is more prevalent: Think of a patient’s chest CT scan and X-ray from a few months apart, or microscopy images and gene expression profiles from different cells of the same type.
Mapping out the mutual information and pairing axes in 2D gives us an intuition of the limits and abilities of a data alchemist.
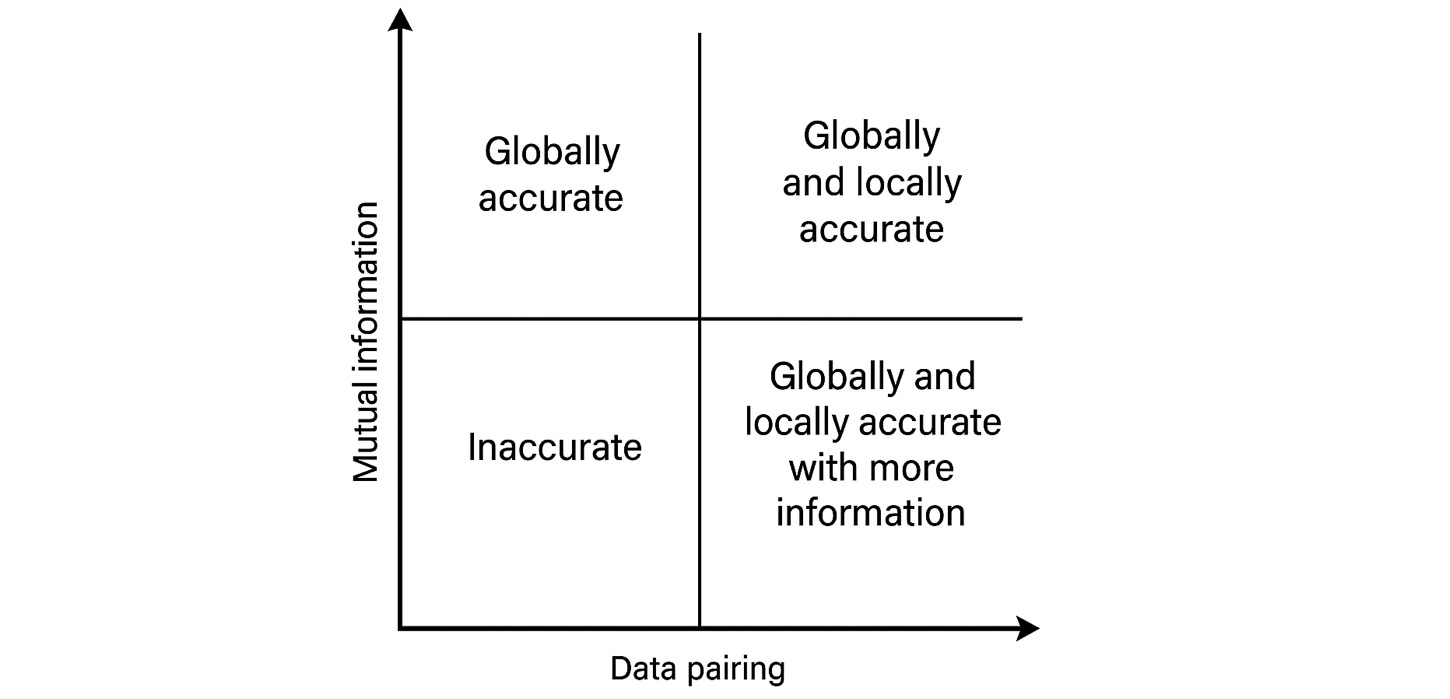
In the upper right, where there is substantial mutual information and excellent pairing, we can expect an AI to seamlessly transform data with high confidence. Moving to the left and decreasing the quality of pairing, a model might correctly infer global content if not exact details – from a patient’s CT scan it could generate an excellent X-ray. The match will not be “one-to-one” with a ground truth, but the underlying biology might be the same.
Decreasing mutual information inherently makes a transformation less certain. Here, strong matching is an advantage if more than two modalities are involved. Consider trying to infer the spatial genomic profiles of a tumor imaged in a histopathology slide. The image itself may say little about certain variants (lower right quadrant). If we fuse sequencing data from a cell free DNA assay (upper left quadrant) with the image, it may permit an educated guess at the spatially resolved profiles. Finally, in the case of low mutual information and weak matching, prospects are slim – suggesting the need for multimodal or higher content protocols.
In practice, it’s hard to ascertain which regime a transformation falls into. To avoid being misled, predictions must come with well-calibrated uncertainty scores. For inferred high-content readouts, a blanket confidence threshold is insufficient, uncertainty will often be unevenly distributed across individual features. If an analysis relies solely on high confidence features, then additional experiments may be unhelpful. AlphaFold exemplifies this with its per-residue scores that help researchers easily identify a predicted structure's strengths and limitations.
Chatting with a data alchemist
Let’s imagine a hypothetical conversation with a data alchemist (generated with the help of o1). Note that “<>” denotes non-text inputs and outputs.
User: Here’s a section of a digital histopathology slide <image> from a patient diagnosed with an early-stage liver tumor. I’d like to see a virtual immunofluorescence image that highlights where Protein A <structure> and Protein B <structure> are expressed. Can you generate that for me?
AI: Certainly! Here is a virtual immunofluorescence image showing the most likely subcellular localization for your proteins of interest: <immunofluorescence image with overlaid confidence maps>.
Some key observations: Protein A (e.g., a cell-cycle regulator) appears concentrated in the nuclei of rapidly dividing cells near the tumor’s core with high confidence. Protein B (e.g., an immune checkpoint marker) localizes on the surface of certain immune cells clustering at the tumor edges; the abundance of this protein is context dependent and hard to infer from morphology alone.
User: This is helpful! Could you also create plausible 3D reconstruction of this region, so I can better visualize the tumor architecture and nearby blood vessels?
AI: Here is the 3D tumor microenvironment model, reconstructed from the 2D image: <3D tumor image with confidence map>. Features to note are:
(1) Tumor Core: A dense cluster of tumor cells consistent with early-stage hepatocellular carcinoma; marked by high proliferation signals. (2) Vascular Network: Small vessels feed the lesion, exhibiting modest signs of new vessel formation. (3) Immune Cells: A moderate population of T-cells is suggested at the boundary, though their exact numbers and activation state vary.
As before, areas where the 2D slide was rich in morphological clues have higher confidence in the 3D model. Less-detailed regions are speculative.
User: I see some immune cells in the periphery. I’d like to isolate one of those cells and generate a predicted single-cell gene expression profile. Let’s check if it matches an activated T-cell phenotype.
AI: Here is the predicted single-cell gene expression for that immune cell: <Single cell gene expression profile>. Summary: (1) Markers like CD8CD8 and IFNG suggest it could be a cytotoxic T-cell. (2) There is also a notable signal for PD−1, an immune checkpoint protein, pointing to partial exhaustion. There is high confidence in canonical T-cell markers (e.g., CD8) and moderate confidence in checkpoint markers, which are context-dependent and influenced by local signaling.
User: I suspect that early blood vessel formation in this tumor is driven by a subset of cells that not only secrete pro-angiogenic factors (molecules that stimulate the formation of new blood vessels) but might also promote immune evasion. Specifically, I think cells near the nascent vasculature express both a vascular growth factor and an immune checkpoint protein. Before I commit resources to a real-world experiment, can you give me a virtual co-staining to see if those markers overlap?
AI: Absolutely! Based on the existing gene expression data, I have generated a predicted co-staining image for a pro-angiogenic factor (e.g., VEGF) and an immune checkpoint protein (e.g., PD-L1) <VEGF_PD-L1_CoStaining_Image>. Preliminary Assessment:
Local Co-Expression: Around 15–20% of cells lining or adjacent to new microvessels appear to co-express both VEGF and PD-L1, suggesting these cells could support blood vessal formation while also dampening local immune activity. Confidence: High in the VEGF signal near newly forming vessels, moderate in the PD-L1 co-expression, since immune checkpoint activation can fluctuate with local signaling cues.
If confirmed experimentally with real co-staining, this subset of cells may play a dual role in promoting tumor blood supply and immune evasion. Such findings could guide targeted interventions—e.g., blocking the growth factor or interrupting the checkpoint pathway.
This example demonstrates the interrogation of data from a single modality. Using the data alchemist to print out latent information in a requested format. If predictions were accurate and uncertainties well-calibrated, the researcher may perform a single experiment instead of 4 or 5. While multimodal generation here was shown to create just one example per request, in practice, having many plausible examples to compare may better reflect the model’s uncertainty than probability scores alone.
Data transformation problems have been studied in other domains
There are parallels to biomedical data transformation tasks in other domains, using diffusion and autoregressive architectures. Basically, it’s a conditional generation problem with multimodal prompts. Text-to-image models illustrate the principle nicely.
A text prompt defines constraints for the image generator to follow. Individual words tend to say little about what’s happening in an image. Short text prompts therefore allow creativity, while long prompts impose additional limits on the generator and shrink its options for valid images. Drawing a connection to biomedicine, high mutual information measurements are like long prompts that describe every minute detail. Text-to-image generation has gotten extremely good in a relatively short time. State-of-the art models, like DALLE, MidJourney, and Stable Diffusion, closely adhere to instructions and produce realistic images.
The success of text-to-image has spawned equally impressive text-to-audio85, text-to-video86, image-to-video87, image-to-3D8889, and multiple text and image prompts to image90. A biomedical data alchemist would take these advances to the extreme with the ability to be prompted with and generate any of dozens of modalities and their combinations.
Creativity is prized in art, but detrimental for scientific data. Quantifying uncertainty and mitigating hallucinations is an open problem. There are rudimentary strategies though. If working with a next-token prediction model, then each token is sampled from a probability distribution that roughly corresponds to uncertainty – albeit not necessarily well-calibrated. Ensembles of predictions, created by querying a model multiple times, can be checked for global “semantic entropy” or local per-token consistency91. Lastly, while fraught, LLMs have some ability to self-evaluate by directly estimating the probability that a statement is true92.
World Model
Simulating observation, intervention, and interaction over diverse time scales
Life is dynamic: Composition, organization, and behavior continuously fluctuate in response to internal and external signals. Measurement snapshots reveal a system’s state at just one slice along a trajectory. Simulating complex biology through time in a digital world would enable identification of regulatory networks and mechanisms and the exploration of treatment outcomes. A world model is a vital piece of a simulator. It specifies the rules and relationships for a system’s evolution, which can be hard-coded based on prior knowledge or learned from data. An AI world model for biomedicine must account for observation, intervention, and interaction.
Observation is forecasting. That is, a dispassionate projection of how a system will progress if undisturbed. It brings static measurements to life – think cellular profiles in an embryo at a single time point being played out as a complete developmental trajectory.
Intervention, on the other hand, studies the consequences of perturbations: If the embryo experiences a change in nutrient availability, how will its development adjust? Counterfactuals, which compare observational and interventional outcomes, are essential for pinpointing causation: Interventions that induce a deviation from the observational path hold causal significance for the deviant traits.
Interaction builds on intervention by modeling subsystems’ influences on each other. A change in one propagates to another and feeds back or forward through a network. For instance, during limb development, signaling molecules create feedback loops that regulate tissue growth. Such changes in one part of the developing limb affect downstream patterning and limb morphology.
Importantly, biomedical events proceed at an array of timescales for observation, intervention, and interaction. The classic example of blood glucose regulation illustrates this perfectly: while glucose binding to cell receptors occurs in microseconds, the insulin response takes minutes, and long-term adaptations like changes in insulin sensitivity unfold over months.
Digitals twins as continuously updated world models
A traditional simulator has a fixed world model. The digital twin is a special kind of simulator that continuously updates its world model with new data. A twin serves as an environment for running observational and interventional simulations; groups of connected twins model interactions.
The state of a twin is available for monitoring or active steering. In monitoring mode, a simulation might show what happens to a patient with or without treatment. Or with a cohort of twins, run an in silico clinical trial for efficacy. Both are undoubtedly useful. Designing a treatment worth testing is even more demanding. In active steering mode, an agent learns to guide a twin’s state to a desired goal. For example, the agent could be a neural network that tries to craft optimal therapeutics and treatment plans to extend the healthspan of a patient. It tests its designs in simulation before returning with an answer.
Twins aren’t just for patients, in fact any biological entity can be digitally replicated, like virtual cells for instance. While for patients our goal is to find interventions that improve health, in cells, the focus shifts to basic questions such as what, why, and how interventions work. High-throughput phenotypic screens, that perturb cells with libraries of genetic and chemical matter, are workhorses in drug discovery. They look for perturbations that revert disease phenotypes in vitro, and use patterns in the screening hits to identify shared mechanisms. The massive search space of perturbations and their combinations is intractable for real experiments, but may not be for virtual ones.
Simulating interactions entails connecting digital twins of multiple entities. For example, with twins from a few organs, we could model the operation of multi-organ axes. These axes are an emerging topic of biomedical research that show the interwoven nature of physiology. Take the heart-brain-liver axis, which defines associations between seemingly disparate phenotypes like heart structure, brain white matter, and liver fat percentage. Knowledge of multi-organ axes is used to isolate upstream disease drivers – potentially, distant from the location of symptoms – as targets for preventative measures. Unfortunately, a systems view of biology like this is underutilized in medicine.
Chatting with a biomedical world model
Let’s go through an example of how world models, digital twins, and simulation fit together. As before, “<>” represents non-text data; additionally “[]” denotes commentary on the internal operation of the model.
User: I’m evaluating a 65-year-old patient who has shown mild cognitive decline over the past two years. I’d like to forecast the progression of this and other symptoms related to Alzheimer’s disease. Here’s the patient’s medical record: <multimodal EHR including brain MRI scans from the past few years, metabolic panel results, APOE4 genetic status, cognitive test scores, and notes about lifestyle factors like diet, exercise, and past medication use>.
AI: [Loading the EHR in the context window initializes a digital twin that reflects the patient’s current physiological and cognitive state, serving as a basis for running informed simulations. The model runs observational simulations of the next five years using knowledge of statistical patterns learned from other patients and its general knowledge of biomedicine]. Here’s a five-year observational forecast for this patient’s cognitive decline, focusing on brain changes, neuroinflammatory markers, and cognitive test trajectories:
Brain Changes: Hippocampal volume shows a ~3–4% annual reduction, slightly above typical healthy aging rates <series: Synthetic future MRI scans (Year 1, Year 3, Year 5) showing progressive atrophy patterns>.
Peripheral Inflammation: CRP (a protein produced by the liver in response to inflammation) and cytokine levels are projected to rise by 10–20% in the next few years, reflecting an overall increase in systemic inflammatory tone.
Brain Microglial Activation: Specialized PET imaging suggests region-specific increases in microglial activation over time. <Projected PET scans (Year 1, Year 3, Year 5) highlighting areas of increasing tracer uptake, indicating elevated neuroinflammation and microglial activity>.
Cognitive test trajectories: Scores on cognitive tests are forecasted to decline by 20-30% <chart: Expected cognitive assessment scores over time with confidence intervals reflecting uncertainty>.
User: Based on this forecast, I think they might be a good candidate for an experimental drug <SMILES string> with a dosage of 50mg twice daily, would it significantly slow disease progression?
AI: [The models reruns the simulation, incorporating the potential effects of the drug, and re-generates the same multimodal outputs as before. It compares the trajectories against the non-intervention baseline]. Yes, this drug may slow disease progression. The effects on amyloid pathology and microglial activation are inconclusive. MRI scans show hippocampal atrophy rate dropping from ~3–4% to ~2–3% annually <updated series of MRI images highlighting differences from untreated results, and chart of estimated hippocampal volumes>. With treatment cognitive test score decline slows by 10–15% compared to no intervention <table: Year-by-year predicted scores under drug + lifestyle vs. baseline>.
User: I just collected additional data regarding cardiovascular health and noticed mild, but worsening, hypertension. How might this hypertension further influence the patient’s cognitive decline?
AI: [Measurements of the patient’s brain are used to instantiate a digital twin for that organ, while updated blood pressure logs and echocardiograms initialize a heart twin]. This mild hypertension is likely to worsen <chart of forecasted weekly blood pressure readings showing a rising elevation> and to contribute to microvascular damage in the brain <MRI scans showing gradually increasing signs of small vessel disease>. This is expected to amplify the hippocampal atrophy rate we saw earlier by 5–10%.
User: Suggest and test strategies to mitigate hypertension that are synergistic with the experimental drug and treatment plan for the neurodegenerative symptoms.
AI: [The model suggests a list of therapeutics and lifestyle modifications for dealing with hypertension, simulates the outcomes, and chooses the one that best meets desired endpoints]. Introducing a low-dose ACE inhibitor such as lisinopril 10 mg daily, a low-sodium diet, and moderate-intensity aerobic exercise at least three times a week may bring systolic blood pressure down by about 10–15 mmHg, reduce inflammation, and improve cerebral perfusion. Hippocampal volume loss slows an additional 5–10% compared to using the Alzheimer’s drug alone. <chart: showing smaller atrophy with controlled BP>.
The simulation process isn’t static, and can be refined with new data. For simplicity this example shows the model generating and summarizing single trajectories. Multiple trajectories would be required to fairly reflect what are sure to be highly uncertain forecasts, especially deeper into the future. While the example cadence of months or years follows standard clinical practice for monitoring a degenerative disease like Alzheimer’s, the correct timescales for optimal simulation are ambiguous.
This example could also have started with the EHR, created an RL agent that generates small molecule therapeutics, and have it learn to actively steer the patient’s digital twin to some optimal health state by searching through chemical space. The AI world model’s state (i.e., “healthiness”) supplies the rewards for training.
World foundation models and complex system simulation in other domains
Observational simulation is similar to video generation. Video generation models implicitly learn physics to realistically portray object motion, fluid flow, collisions, and textured surface interfaces9394. No model flawlessly adheres to physics, yet it turns out that observing the world through millions of hours of video leads to remarkably good approximations. Likewise, temporally resolved biomedical data, equivalent to videos, might inform the learning of the dynamics of biological systems.
A fully realized world model supports interventions too. Cosmos is a world foundation model combining video generation and an encoder for perturbations (e.g., text descriptions, robot actions, and others)95. It's designed to be a digital twin for the real world, within which robotic agents test and learn behaviors that transfer outside the simulation. Genie 2 is another world model that simulates diverse virtual worlds instead of the real one96. The setting for each world is initialized with a single frame created by a text-to-image diffusion model. Trained at scale, Genie 2 shows emergent capabilities for “object interactions, complex character animation, physics, and the ability to model and thus predict the behavior of other agents”. Applying the same idea to medicine, swap the single frame image used to initialize the virtual world for a health record as in our chat example.
Simulators and world models trained on videos have the advantage that Newtonian mechanics is well-behaved. Biology is not. The weather is a better analogy. GenCast is an autoregressive diffusion model that creates 15-day weather forecasts more accurately and much faster than the best classical techniques97. Ensembles of predictions give well-calibrated confidence intervals and show the spectrum of potential trajectories for extreme weather events like hurricanes and cyclones.
However, there are features of biomedicine unaccounted for in other domains. First, there is incomplete observability with only imperfect partial views of internal state. Second, there are no dense recordings of parameters for a single human like there are for Earth’s weather. A world model would have to learn from measurements acquired from a multitude of entities and somehow piece together general principles of operation. Third, biological systems have agency, while physical systems do not. Perhaps, this makes world modeling easier, assuming an understanding of intentions lessens the requirement for a complete mechanistic understanding (more on this later).
Translational Reasoner
Therapeutic hypotheses and rational drug design
Translational reasoning applies knowledge of phenomena in one context or level of organization to explain observations elsewhere. It’s empirically grounded storytelling. Consider how we might write a disease's story: from a genetic mutation altering protein structure, through its effects on cellular pathways, to tissue dysfunction, and ultimately to the physiological symptoms experienced by a patient. This explanation adopts human-constructed abstractions rather than nature's own logic. Cells don't think in terms of "pathways" and genes don't conceive of themselves as "code for proteins". Good conceptual frameworks make biology comprehensible without straying too far from reality or sacrificing too much predictive power. Practical utility outweighs metaphysical truth.
The stories we tell about disease guide therapeutic hypotheses – proposals for how to disrupt disease mechanisms and promote health. Say we believe a normal protein is overactive and driving cancer progression (as is the case with certain tyrosine kinases). That naturally suggests a solution: use an inhibitor to reduce its activity. This strategy, called rational design, is logical and has worked great for treating a range of diseases.
Distilled to the bare essentials, drug discovery proceeds in four steps. First, generate hypotheses about the cause of the disease. Second, use knowledge of biology to reason about each hypothesis and select the one that best explains observations. Third, design a therapeutic to address the root cause or disrupt its downstream effects. Fourth, test it in the process of a clinical trial. The first two steps of creating and thinking through hypotheses are fundamental, yet often stymied by the labyrinthine causal structure of biology, which defies straightforward explanations.
The multiscale searching for disease causes
A popular and successful procedure for formulating causal hypotheses is the genome-wide association study (GWAS). GWAS comb through genetic data from large populations to identify single nucleotide variants associated with a trait (e.g., disease status, height, BMI, etc.). Statistically significant associations are leads for investigating genetic contributions to disease. GWAS is inherently multiscale, aiming to establish a connection between DNA, one of the lowest levels of organization, and gross phenotypes apparent at the level of organs and organisms. There’s no guarantee that an association indicates a causal relationship, but it may. Literature, in vitro experiments, and animal studies build the case for and against.
The reductionist assumption underlying GWAS is that genes themselves are significant causal factors for disease. But reality is much richer.
The genome is a toolbox that cells use, it isn’t a code that controls their destiny. A fraction of genetic tools exclusively fill a niche, variants that impede their function are detrimental. Indeed, they are primary drivers of monogenic diseases. The majority of tools in the genome are replaceable (compensation), synergistic (epistasis), and contingent (disordered proteins). Most genes, on their own, have minimal influence on physiology. With changes in just one showing slight and nuanced consequences, it may take the wrong combination of dozens or hundreds to raise the likelihood of disease (polygenic risk). Diseases may have no strong genetic component at all.
GWAS is constrained by its focus on associations between just two levels of description: gene variants and high-level traits. While it can point us toward relevant genetic loci, it doesn’t explain how or why those variants influence disease. In truth, causality may reside anywhere in the biological hierarchy, from the subcellular to the organ level. What if we systematically scanned for associations across all relevant strata—such as patterns in cellular pathways, transcriptional profiles in specific cell types, or even proteomic shifts in different tissues? One recent method98 moves in this direction by incorporating knowledge graphs. It aggregates genetic evidence to detect subtle variant effects and uncovers deeper links to cell types and pathways, hinting at how we might move beyond raw correlations to mechanisms.
A powerful translational reasoner constructs causal explanations that bridge multiple levels of biological organization. Rather than stopping at “genes associated with trait X,” it examines how an alteration in gene expression or protein function triggers a cascade of cellular events, culminating in tissue-level pathology and clinical symptoms. By filling in these mechanistic gaps, which GWAS alone can’t provide, translational reasoning supports rational drug design—clearly indicating where therapeutic intervention might most effectively disrupt the disease. The following conversation demonstrates how an AI-driven translational reasoner weaves multiscale causal stories into plausible and actionable strategies for personalized medicine.
Chatting with a translational reasoner
This example will differ from the others in that the reasoning process isn’t suitable for a back-and-forth chat. We’ll start with a prompt and watch the AI’s internal monologue:
User: Here’s the full electronic health record for a patient recently diagnosed with rheumatoid arthritis. What’s the most probable explanation for how the disease developed given their profile? <Attached multimodal health record>
AI: I have been given a multimodal EHR for a patient with newly diagnosed rheumatoid arthritis (RA). The record includes: Standard labs, imaging (hand/wrist MRI, partial ultrasound scans), lifestyle and clinical notes describing symptom onset, comorbidities, and family history.
My objective is to formulate a causal explanation of why this patient’s RA arose – moving from the local, immediate mechanisms (joint inflammation) toward deeper, upstream drivers. I can generate extra data (Data Alchemist style) and use a World Model to evaluate each hypothesis.
Step 1: Identify Proximate Cause
Initial Hypothesis: The immediate cause of painful, swollen joints is infiltration of immune cells into the synovium (connective tissue that lines the inner surface of joints), leading to chronic inflammation <hypothesized white blood cell (WBC) counts in synovial fluid and MRI showing synovial thickening assuming this is true>.
[The model runs two evaluations for the hypothesis. First, it rolls back to an earlier time in the patient’s EHR-based digital twin and inserts the hypothesized measurements. It checks in simulation if that does in fact lead to RA symptoms. This verifies that the hypothesis is explanatory. Second, it compares the hypothesized measurements to the actual ones observed for the patient. That serves to validate that the hypothesis is plausible for the given patient].
The EHR’s lab results show elevated WBC, and the MRI shows pronounced synovial hyperintensity around the joints. This strongly supports local inflammatory infiltration. Conclusion: Proximate cause is confirmed with high confidence (~90%).
Step 2: Grow the Chain Deeper: Which Immune Subsets Are Key?
Having established that infiltration is the immediate driver, I explore the next level: which immune cells or signals trigger the infiltration?
Hypothesis 1: Innate immune activation (e.g., macrophages, neutrophils) responding to an unknown stimulus <hypothesized single cell RNA-seq dataset showing high expression of IGKC and PRDM1 in B cells with uncertainty>
Hypothesis 2: T- and B-cell cross-talk, featuring auto-reactive T-helper cells and plasma B-cells producing autoantibodies <hypothesized single cell RNA-seq dataset showing high expression of IFNG, IL17A, and TBX21 in T cells with uncertainty>
[As before the model checks the hypotheses for explanatory power and patient relevance. In this case, it uses data alchemy conditioned on actual data to generate B cell and T cell expression profiles with appropriate uncertainty estimates].
40% of simulated trajectories under hypothesis 2 end with the observation of RA symptoms compared to 20% under hypothesis 1, and 1% under a null hypothesis. Expression profiles theorized under hypothesis 2 align better with expectations for the patient based on lab results of synovial fluid, blood, and imaging studies. Conclusion: Next-level cause is likely T/B-cell–mediated autoimmunity. Confidence: ~25-30%.
Step 3: Explore Further Upstream: Autoimmune Triggers
Next, I ask: Why are T-cells and B-cells so reactive? Potential deeper explanations:
Hypothesis 1: Autoimmune Regulator (AIRE) protein is mislocalized or low in thymic tissue <hypothesized H&E biopsy slide with spatial proteomics for AIRE showing low abundance>
Hypothesis 2: Microbiome imbalance triggers cross-reactive T-cells (molecular mimicry) <hypothesized gut metagenomic profiles showing an overrepresentation of Prevotella copri>
[The AI adds the generated data to the patient’s digital twin and runs a multi-year simulation to see whether these hypotheses lead to the reactive T-cells and B-cells gene expression profiles anticipated in the previous step. If not, it may propose and test other hypotheses or backtrack].
About 30% of the simulated trajectories under Hypothesis 2 replicate the specific patterns of synovitis, autoantibody production, and T/B hyperactivity found in the patient’s real data. In contrast, Hypothesis 1 drives autoimmunity in only ~5–10% of trajectories and is less consistent with the patient’s history.
Conclusion: The overrepresentation of Prevotella copri is the more likely upstream explanation. Confidence: ~30%. Thymic anomalies may still play a role, but the simpler, higher-confidence story is that a microbiome imbalance primes T- and B-cells to mount an autoimmune attack on joint tissues.
Step 4: Decide Whether to Go Deeper or Conclude
I could keep going—investigating environmental triggers (e.g., past infections) or genetic predispositions. However, I evaluate:
Explanatory Sufficiency: The identified gut microbiome imbalance plus local infiltration by autoreactive T/B-cells sufficiently explains the chronic inflammatory loop in the synovium.
Actionability: Targeting deeper-level factors are unlikely to change the immediate therapeutic approach.
Uncertainty Escalation: Additional layers add speculative data with broader error bars.
Hence, I conclude that I have a plausible and clinically relevant causal chain: gut microbiome imbalance → autoreactive T/B-cells → synovial inflammation → RA symptoms.
Admittedly, this is aspirational and only an illustrative toy example of what would unavoidably be a messy and uncertain reasoning process. Some limitations are:
This example assumes a single primary driver, whereas in reality disease arises from a confluence of genetic and environmental factors. Modern medical practice struggles to account for all these influences too.
Later I’ll make the case that it might not be insane, but inferring advanced “-omics” data (e.g., single-cell gene expression or gut microbiome profiles) purely from standard EHR information is improbable. A more realistic strategy could draw on multimodal data from other patients or experiments to perform retrieval augmented generation (RAG).
In the same vein, the dialogue doesn’t describe plans for confirmatory lab work or specialized imaging to test hypotheses. In practical settings, physicians or researchers would order panels (e.g., autoantibody tests, stool microbiome analyses) to gather direct evidence. Without the right data, confidence estimates for any one hypothesis are likely to be low and will degrade with the length of the causal chain. This is partly why I proposed having separate evaluations for explanatory power and P(True|Patient). The first is agnostic to available patient data while the other depends on it.
Deep research of literature is another valuable metric for hypothesis evaluation not shown here.
Unless counterfactuals are robustly tested and the simulator is well-calibrated, there is no guarantee that the proposed chains are truly causal. What seems like a mechanism might simply be an association.
Despite limitations and uncertainties about how a translational reasoner would work, the value is evident. Instead of piecemeal or purely correlative studies, it offers up a complete therapeutic hypothesis, potential biomarkers and therapeutic targets, and a testable research plan.
Reasoning at the research frontier
Frontier research gives us a modicum of hope that translational reasoning isn’t a pipe dream. Especially, the success of reasoning models leveraging structured search, reinforcement learning, and multi-agent systems.
Monte Carlo Tree Search (MCTS), which inspired the example above, is the basis for AlphaGo99. It iteratively selects and evaluates game moves, analogous to the selection and evaluation of hypotheses. MuZero extends AlphaGo by learning allowed actions and evaluating them using a neural network, without access to an exact game simulator100. Likewise, in biomedicine, we do not have a deterministic simulator of states and responses, but we might be able to learn a data-driven world model. Closer to our intended use case ReST-MCTS* applies MCTS to LLMs, assigning rewards to the steps in a chain-of-thought based on heuristic quality, distance from the correct answer, and other factors101.
State-of-the-art reasoning LLMs are trained with reinforcement learning and employ test-time scaling, without structured search, to tackle hard questions. Pioneered by OpenAI’s o1, they create long chains-of-thought that explore solution space before settling on an answer102. During training, they organically learn to evaluate thoughts on their likelihood of leading to the answer, continue promising thoughts, and backtrack at deadends. While we don’t have specifics for OpenAI’s models, DeepSeek R1 shows similar reasoning abilities103. R1 incentivizes learning with heuristic rewards like proper format, applicability to the question, and correctness. Training initially emphasizes verifiable domains like math and coding, but later incorporates supervised finetuning for coherence and broader reasoning abilities.
The AI co-scientist is an exciting system that brings sophisticated LLM agents to biomedicine104. The co-scientist is a multi-agent framework for generating, evaluating, and refining biomedical hypotheses. Prompted by a research goal from a scientist, the agents collaborate to find an answer that is “plausible, novel, testable, and safe.” To achieve this, the system employs structured self-critique and debate mechanisms, where specialized agents rigorously argue, defend, and refine hypotheses. A key element of this process is the head-to-head tournament ranking system, which uses an Elo-based framework to rate and improve competing hypotheses. As in our example, part of the evaluation process involves a Reflection agent, which tries to simulate the outcomes of proposed experiments. This preemptively identifies weaknesses before real-world testing. It has characteristics of a translational reasoner though it isn’t multimodal.
Whether reasoning is explicitly structured with MCTS, developed by reinforcement learning, or the product of multi-agent debates, these methods are new and rapidly advancing. Multimodal reasoning is at an even earlier stage. LLaVA-CoT trains a multimodal LLM to analyze and answer questions about images by following a pre-defined reasoning template105. The model reasons about images but does not generate them. Whiteboard-of-thought lets an LLM write and run code for data visualizations, feeding the plots back to the model to enhance its thinking106. Breakthroughs are necessary to support multimodal translational reasoning, especially when there are dozens of modalities to work with.
Although not explicitly designed for causal modeling, one study found that LLMs surpass the best performing models designed for causal inference on pairwise causal discovery (e.g., does A cause B) and counterfactual reasoning tasks107. The extent to which this constitutes true causal understanding is debated, yet LLMs prove valuable in coming up with plausible hypotheses. Moreover, theoretical work has shown that causal structure may be learned from “passive data [when it] contains the results of experiments”108. Illuminating these causal structures across scales is precisely what a translational reasoner must achieve to propose accurate, mechanism-based therapeutic hypotheses.
Prospects
Having outlined how an AI data alchemist, world model, and translational reasoner could reshape biomedical research, we now must ask just how ambitious these ideas are. They would demand robust causal inference, multimodal synthesis, and intricate simulations of complex dynamical systems—capabilities none of today’s models fully deliver. The AI frontier offers tantalizing glimpses of these abilities in other domains, and I see a faint through line from what is currently possible to what powerful biomedical AI might achieve by building on, scaling up, and refining these foundations. Still, skepticism is justified.
In this section, I’ll address what I see as the biggest obstacles and causes for concern. First, briefly touching on technical and engineering aspects before turning to the scientific questions around data quality, quantity, and format (i.e., how information is structured and presented for learning).
Technical Challenges
Tokenizing and training on all biological data ever collected would be a feat. But compute resources probably won’t be a limiting factor. 100,000+ GPU clusters are becoming standard and breakthroughs like DeepSeek-V3 show that clever engineering gets similar results with much less109.
On the modeling side, a few important areas saw significant progress in the last year. There are good tokenizers for most biomedical data types – sequences, 3D structures, images110, gene expression, videos111, and time-series112. Newer schemes using byte-level encoding to work with raw data may circumvent the tokenization altogether113.
2024 marked the release of the first really scalable, performant, and open-source AMMs for unified multimodal generation. Further maturation is inevitable, and it’s a foregone conclusion that AMMs will soon be the dominant paradigm. With internet text exhausted, other modalities facilitate continued scaling of pre-training and consolidate modality-specific models and tasks. Autoregressive reasoning models are in their infancy, but already show breakthrough performance for math and science.
Long-context reasoning on hundreds of high-dimensional multimodal readouts, as might be required for a translational reasoner, would represent a major leap. Fortunately, context windows are on an upward trajectory. GPT-3.5 had a window of under 20K tokens when released in late 2022, GPT-4 reached 128K in 2023, Gemini hit 2 million tokens and then 10 million tokens both in 2024114.
If treating every nucleotide in DNA as a token, there’s a way to go before handling a whole genome sequence. Architectures that use a “neural memory” to memorize and compress previous tokens are one option115. As are efficient tokenizers that group redundant features116 or concepts117. Or maybe we skirt the issue by operating directly in the space of continuous latent tokens to avoid token-heavy intermediate outputs118.
I’ll concede that this analysis is a bit dismissive of what are substantial engineering barriers. I’m optimistic about the creativity and resourcefulness of AI scientists and engineers to continue the breakneck pace of innovation. Truckloads of investor money and the ever-decreasing cost of compute help too.
Data Challenges
The upper bound for an AI system is set by its training data. Biomedical data has quality problems: it’s rife with confounders, batch effects, noise, errors, incommensurate and non-reproducible results, and the absence of critical metadata. More troubling, the data we have is shaped by what we know how to measure – it is a skewed representation of biology. We settle for 2D histopathology images to analyze tissue that is 3D, record single snapshots of gene expression in living organisms, and depend on natural experiments and observational studies to analyze human genetics.
Data isn’t in the appropriate format for training a powerful AMM either. Inculcating multimodal integration and multiscale reasoning abilities necessarily means having access to documents containing multimodal and multiscale data. In the review, we saw models trained on various arrangements of interleaved text, protein sequences, small molecules, and DNA. They mark early efforts in the right direction, but are insufficient for the much grander scope of our objectives.
Finally, measured in bytes, quantity may not appear to be an issue, but biology is extremely high-dimensional. Consider that a human genome has 3 billion base pairs, nearly every human has a unique one, and a single “wrong” nucleotide can make the difference between health and disease. Environment profoundly affects health too, substantially more than genes do119. The “curse of dimensionality” raises doubts about whether the rules governing biology are learnable from finite data120. Biomedical state space may be so enormous that data-driven simulators and reasoning processes cannot skillfully interpolate outcomes in unseen cases.
I don’t have solutions for these problems aside from waiting for better data and experimental techniques (or lab agents121122123). Yet, I doubt things are as dire as they seem. First, I’ll propose ways to organize our imperfect data to make the most of it – hopefully, alleviating quality and format concerns while bringing us closer to data alchemists, world models, and translational reasoners. Second, I'll suggest that the complexity of biomedicine may not demand the overwhelming volume of data one might initially assume.
Building the training dataset
Naively tokenizing existing datasets and feeding them to an AMM would result in a model that processes biomedical modalities with shared weights, yet fails to fuse information seamlessly. Akin to a multilingual LLM that knows many languages in isolation but cannot translate between them or follow a mixed-language conversation. That’s an unsatisfying outcome. Research boundaries like basic vs. clinical or genetic vs. cellular exist for convenience, not because they reflect fundamental divisions. Biological processes never exist in isolation; genes, cells, and organs co-evolve and co-regulate through complex interaction networks. AI that segregates domains will miss the essential interconnected nature of biological systems.
Instead, training data should consist of documents that encapsulate our observations of biological phenomena across modalities and resolutions. For this we look to scientific publications and LLM-augmented data curation.
Scientific publications
The best publications are integrative by nature in that they weave together lines of evidence from multiple experimental assays to prove a point. The narrative is coherent, usually multimodal, and fashioned for human interpretability.
In the review section, we covered methods that use named entity recognition to replace small molecule and protein names in papers with actual structures and sequences. The same could be done for other modalities. Take for example a paper that links an upregulated gene in a cell type to a disease. Assuming the observation of the upregulated gene was derived from single cell transcriptomics, the raw data supporting the authors' conclusion is inserted in the proper section of the text. Embedding data in publications supports learning from the “Methods” section too. A model might discern features in the data that are spurious and caused by quirks in an experimental process. In this way, contextualizing data perhaps mitigates the influence of confounders, batch effects, and other quality concerns.
Unfortunately, publications are manicured science. They present positive results and gloss over the messy business of failed hypotheses and experiments common in research. To avoid those pitfalls, they ought to be included in the dataset. Besides, there are repositories of biomedical data stored in research databases and biobanks not yet studied in literature.
LLM-augmented data curation
LLMs, which have impressive knowledge of biomedicine, may help to curate dissociated data into solid multimodal and multiscale documents. To make this concrete, let’s work through a hypothetical pipeline (also see the Appendix for examples):
1. Randomly choose a document context from an ontology. The context defines the overarching topic, perhaps a gene, protein, person, disease, animal species, etc. Level of granularity is adjustable because of the hierarchical structure of ontologies. Organizing data under an aligning context reduces the need for explicit pairing. Narrow contexts increase the odds that tokens in one modality will be predictive of tokens in another, which is the basis for multimodal fusion. Broader contexts are helpful for discovering unexpected relationships.
As an extreme case demonstrating the importance of context, imagine a model trying to predict a protein structure from a PET scan of a brain. Absurd, right? What if the protein in question is amyloid and the PET scan is of a brain showing amyloid plaques? Certainly, the PET scan doesn’t contain enough detail to predict the full structure, but it might predict a non-zero fraction. In this case, maybe the presence of aggregation-prone structural motifs. Learning connections from brain imaging to protein structure is supported by the unifying context of Alzheimer’s disease – we’re encouraging data alchemy.
2. Pick a template type. The template specifies the order of data. Crafted either by a human or dynamically by an LLM. Logical ordering (i.e., low perplexity) matters for autoregressive models because they are sensitive to the arrangement of sequences. Some organizing principles are temporal, top-down, and bottom-up.
A patient’s medical history or a pseudotime (i.e., separate cells but the same process) developmental trajectory, for example, are temporally ordered from oldest to newest. Interventional data follows the same format where “before” corresponds to unperturbed and “after” is perturbed. Both support world modeling and simulation.
Top-down and bottom-up describe levels of organization, where a top-down document orders data from organism-level to the molecular-level and bottom-up does the reverse. Modalities that describe the same time point or level may be ordered randomly or based on prior knowledge about the system (e.g., chromatin accessibility proceeds gene expression). Sequentially arranging data by level of abstraction supports translational reasoning.
As a special case, supervised tasks rely on explicit input-target sequence pairs. These pairs are concatenated as standalone documents or inserted as sub-documents. For example, in a protein folding task, sequence and structure may be placed within a broader document that describes processes where the protein is involved, thereby contextualizing folding in a system.
3. Fill the template. An LLM decides from text metadata if a readout fits. Beforehand, missing and non-canonical metadata would be imputed and standardized by specialist multimodal chatbots such as those for clinical images. Frontier models are competent with biomedical texts124. LLM perplexity on neuroscience abstracts, for instance, is a good predictor of whether the research is real or bogus – better even than professional neuroscientists125. This suggests they might skillfully decide which data makes sense together in general. Encouraging the selection process to incorporate data from distinct experiments may additionally boost robustness to noise and covariates, as nuisance information will not be consistently predictive throughout the document.
4. Interleave multimodal data with text. An LLM adds text conditioned on metadata, drawing on its significant knowledge of biomedicine learned from reading literature. When available, experimental methods are placed alongside the actual readouts.
Scientific publications and curated documents make for a compelling multimodal and multiscale pre-training dataset. To recap, data alchemy has the opportunity to arise from ground truth input-target pairs and multimodal data that’s aligned by context. World modeling is encouraged with temporally ordered and interventional data. Translational reasoning is motivated by scientific publications and hierarchically ordered data.
Post-training, including supervised finetuning and reinforcement learning, serves to further amplify these embryonic modes of intelligence. I won’t cover post-training other than to note that professional materials are a good resource for alignment. Medical guidelines from recognized bodies set normative frameworks for clinical decision-making. Evidence-based reviews demonstrate proper evaluation of conflicting evidence. Case reports illustrate how abstract principles apply to specific patients. Adhering to the format of these gold-standards by including caveats, confidence levels, and citations should be rewarded.
Curse of dimensionality
Regardless of how cleverly the data is arranged, no new information is ever added. New basic research and clinical data is digitized and banked every year. Does it encompass enough knowledge to realize powerful biomedical AI? That’s unanswerable from first principles and it depends on the precise capability and task. Despite not knowing exactly how far away we are, I believe that we’re closer than one might expect. Not necessarily close, just closer.
Hidden latent information
Our brains are hardwired with an instinct for language and parsing natural images. Not so for DNA sequences or clinical images – interpreting those patterns is a hard-won skill after years of study. AI systems start fresh, with minimal implicit bias to overcome. The result is striking: they routinely see more patterns in biomedical data than human experts. Let’s recap examples from the review section and elsewhere:
Deep neural networks analyzing routine eye scans predict biological sex, age, smoking status, blood pressure, and cardiac risk – features "not previously thought to be present or quantifiable in retinal images"126127.
A language model predicts protein localization within cells using only amino acid sequence information, proving that protein sequences themselves have unexpected information about subcellular distribution.
AI models, from CNNs to self-supervised vision transformers, can predict molecular characteristics and patient outcomes directly from standard H&E-stained slides128. These models extract morphological patterns too subtle for human pathologists to consistently recognize.
SSL on wearable device signals can determine demographics, diagnosed conditions, and medication usage as well as foresee health events like heart failure. Simple regression models, trained on accelerometry data, detect subtle charactertistics of Parksinon’s disease years before diagnosis129.
Autoregressive models trained on continuous glucose monitoring data internalize signatures of liver, kidney, and cardiovascular health.
Standard chest X-rays show subtle indicators of cardiovascular disease risk and diabetes, enabling “opportunistic” screening without additional tests130.
These discoveries defy a compartmentalized view of biology. They aren't coincidental correlations but evidence of an interconnected biological network that AI can detect because it is unconstrained by categorical thinking. An organism is a single system where information flows across the traditional boundaries we've imposed for analytical convenience.
The long recognized complexity of biology has driven the embrace of high-dimensional readouts like omics and images. Consequently, vast repositories of untapped information exist within the data we've already collected. Connecting back to an earlier discussion, while humans need data to be structured in specific ways for interpretation, AI systems extract meaning anywhere that a signal is present. Gaps in interpretation do not imply gaps in data coverage.
Dimensionality reduction
Biological systems face an immense state space. Consider the countless ways in which 20,000 genes, 80,000 proteins, and 37 trillion cells could interact to produce novel forms and functions. Viewed as a random collection of biomolecules, we might expect life to be chaotic and unpredictable. Oddly, we observe order: reliable development, homeostasis, and adaptability. How does life rise above the curse of dimensionality to achieve such stability? Through dimensionality reduction – the compression of state space onto lower-dimensional manifolds.
Reduction emerges because life pursues goals. A cell doesn't randomly explore all possible states – it actively regulates its internal environment within confined parameters (optimal pH, ion balance, etc.). Homeostatic setpoints are attractor states; magnets that pull a system back when it deviates131. When cells organize into tissues and organs, they form networks pursuing collective objectives like the retention of shape and normal function. Only a modest fraction of physical states are compatible with life. The persistence of organisms in these life-supporting states, driven by natural selection, explains the existence of the ordered systems we recognize as living things. Examples of dimensionality reduction appear throughout biology.
At the molecular level, proteins could theoretically adopt an astronomical number of conformations (Levinthal's paradox). Yet they consistently fold into functional structures by following restricted energetic trajectories toward low-energy states.
At the cellular level, humans have only a few hundred cell types characterized by their gene expression profiles (usually they are clearly visible in just 2 UMAP dimensions!). During development, stem cells follow "canalized" trajectories toward these predetermined cell fates132. The molecular circuitry within cells and signals from their environment sharply curtail divergence.
At the organism level, convergent evolution demonstrates how unrelated species independently evolve similar traits when facing comparable environmental challenges – for instance, camera-like eyes in vertebrates, cephalopods, and some jellyfish. Selection pressures channel evolution toward a bounded set of functional outcomes.
Finally, when self-regulation fails, we observe a finite set of dysfunctions. Cancer illustrates this principle. The genomic changes behind malignancy include countless mutations, cancers show just ten fundamental "hallmarks," including sustained proliferation and immune evasion133. These hallmarks represent a compressed space of phenotypes necessary for cancer cells to survive. Diseases themselves can be understood as attractor states within physiology's dynamical landscape, explaining why a single disease, as defined by symptoms, may come about from various distinct causes134135. Different starting points converge on the same outcome whether in health or disease.
In essence, the same principle that renders biological systems tractable—compressing themselves onto a tiny subset of state space—also makes them amenable to AI. Systems governed by homeostatic or functional goals cluster around stable attractors. Instead of wasting effort enumerating the astronomical possibilities of a high-dimensional space, an AI can focus on identifying which attractors exist, mapping how the system transitions among them, and learning the shape of that manifold. Understanding that biology generally sticks to a confined set of viable trajectories, may make AI analysis and forecasting in this complex domain feasible.
Conclusion
I was inspired to write this essay because I felt there was a disconnect between frontier and biomedical AI research. On the frontier side, Machines of Loving Grace explored what AI might revolutionize in the biological sciences, but was vague on how. The author projected that powerful AI will come as early as 2026 – sprouting from the likely conviction that the current trajectory of AMMs trained to reason with reinforcement learning and self-play will get us there.
On the biomedical side, I haven’t seen anyone take the idea of large autoregressive models seriously for anything other than text. How to build the virtual cell with AI136 and Toward AI-Driven Digital Organism137 are insightful and principled perspectives on how to think about and model the multiscale structure of biological systems. Yet, the “bitter lesson” looms in the background138. Autoregressive models are not a natural fit for biomedicine – but they are scalable and unencumbered by human assumptions about the data. I thought it worthwhile to investigate what the convergence of frontier AI research and biomedicine could look like in the coming years and what we should hope for it to accomplish.I acknowledge that biomedicine is a grand challenge in science that won’t be “solved” by throwing data at a huge model. But would having a model like that make a meaningful difference in research? Hopefully, this essay has made a compelling case that the answer is yes.
Do I think it could be built in the next 10 years? A year ago I would have said no. Back then, we didn’t have performant AMMs, long context windows, world foundation models, research agents, or reasoning models. Progress on the exponential curve is unforeseeable. Now, I say maybe.
Postscript: The scaling law for biomedicine
One final speculation (if you’ll indulge me!): scaling laws for LLMs are empirical relationships that correlate benchmark performance with increased compute (i.e., number of parameters, training tokens, and weight updates). I propose a corollary for biomedicine: providing AI models with system-level context will spark emergent capabilities, especially for problems where a single-modality approach has failed. Patterns that are invisible when a domain is siloed will become obvious when assimilated with other basic and clinical data – even when we aren’t quite sure how they relate. Underscoring a central lesson of systems biology, that “the parts cannot be understood except in the light of the whole”.
Appendix
What follows is a collection of sample LLM-curated documents that we might construct for use in a pre-training dataset. All generated with the help of Claude 3.5 Sonnet.
Patient document
The first document type is centered on a single patient and looks like a standard medical history. Data is ordered temporally.
<Demographic> DOB: 05/15/1955 (69 years old) Sex: Male Race: Caucasian Smoking history: 40 pack-years (quit 2 years ago) Occupation: Retired construction worker
<Notes> Patient presents with persistent cough for 3 months, fatigue, and recent weight loss of 15 lbs. Physical examination reveals decreased breath sounds in the right upper lobe. Recommending chest X-ray and complete blood count.
<Lab test> Complete Blood Count (CBC): WBC: 11.2 x 10^9/L (High) RBC: 4.1 x 10^12/L (Low) Hemoglobin: 11.8 g/dL (Low) Hematocrit: 35.4% (Low) Platelets: 385 x 10^9/L (High normal)
<Chest X-ray Study and Diagnostic report> Chest X-ray reveals a 3.5 cm opacity in the right upper lobe. Mediastinal lymphadenopathy is also noted. Findings are suspicious for primary lung malignancy. Recommend CT-guided biopsy for definitive diagnosis.
<Notes> Discussed X-ray findings with patient. Explaining the need for a biopsy to confirm diagnosis. Patient agrees to proceed with CT-guided biopsy of the lung mass. Scheduling biopsy and ordering additional blood tests, including tumor markers.
<Histopathology Study and Diagnostic report> Microscopic examination reveals poorly differentiated non-small cell carcinoma, favoring adenocarcinoma. Immunohistochemistry positive for TTF-1 and Napsin A, confirming lung adenocarcinoma. Molecular testing shows EGFR exon 19 deletion mutation.
<Notes> Diagnosis: Stage IIIA (T2N2M0) EGFR-mutated lung adenocarcinoma. Treatment plan: Initiating targeted therapy with osimertinib (Tagrisso), an EGFR tyrosine kinase inhibitor, at 80 mg orally once daily. Will monitor for side effects and assess response after 2 months.
<CT imaging study, lab test, and notes> After 2 months of treatment: CT scan shows 50% reduction in primary tumor size and decreased mediastinal lymphadenopathy. CEA tumor marker decreased from 15.2 ng/mL to 4.8 ng/mL (normal range: 0-3 ng/mL). CBC shows improvement: WBC 8.1 x 10^9/L, Hemoglobin 13.2 g/dL. Patient reports improved energy levels and resolution of cough.
Besides the inclusion of three distinct imaging studies and physicians notes, there are opportunities to replace a few gene and small molecule names with DNA sequences, protein structures, and SMILES strings as well.
Phenotype (disease) document
In a phenotype/disease document, we center data on an observed clinical trait. Unlike a patient document, we can mix and match records from anyone that has been diagnosed. In this example, we order modalities top-down:
<Description> HER2-positive breast cancer is an aggressive form of breast cancer characterized by overexpression of the human epidermal growth factor receptor 2 (HER2) protein. This subtype accounts for approximately 20% of all breast cancers. HER2-positive tumors tend to grow and spread more rapidly than other breast cancer types, leading to a poorer prognosis if left untreated.
<MRI from patient 1> [MRI Image] T1-weighted contrast-enhanced MRI of the left breast shows a 2.8 cm irregular mass with spiculated margins in the upper outer quadrant. The mass demonstrates rapid enhancement with washout kinetics, typical of malignancy. Axillary lymph nodes appear enlarged, suggesting potential metastatic involvement.
<Histopathology image from patient 2> [Histopathology image] H&E stained section at 400x magnification reveals sheets and clusters of pleomorphic tumor cells with prominent nucleoli and frequent mitotic figures. The cells show a high nuclear-to-cytoplasmic ratio and form occasional glandular structures. Immunohistochemistry demonstrates strong, complete membrane staining for HER2 in >10% of tumor cells (3+ score), confirming HER2 positivity.
<Epigenetics profile from patient 3> [Epigenomics profile] Analysis of the DNA methylation array data reveals distinct methylation patterns associated with HER2-positive breast cancer. Hypermethylation is observed in several tumor suppressor genes, including PTEN and CDKN2A. Conversely, hypomethylation is noted in regions associated with ERBB2 and genes involved in cell cycle regulation. The overall methylation profile shows significant divergence from normal breast tissue, with a global hypomethylation pattern characteristic of many cancers.
<Bulk transcriptomic profile of patient 4> [Transcriptomic profile] RNA-seq analysis shows high expression of ERBB2 and other genes in the HER2 signaling pathway, including GRB7 and STARD3. Upregulation of genes involved in cell proliferation (MKI67, CCNB1) and anti-apoptotic pathways (BCL2, BIRC5) is also observed.
<Sequences of known causal gene variants> [Genetic sequence] TP53 mutation: c.524G>A (p.Arg175His) - A common missense mutation affecting the DNA-binding domain of p53. [Genetic sequence] PIK3CA mutation: c.3140A>G (p.His1047Arg) - A hotspot mutation in the catalytic domain of PI3K, leading to constitutive activation. [Genetic sequence] PTEN deletion: Large genomic deletion encompassing exons 2-5, resulting in loss of PTEN function.
One challenge for this document type is that phenotypes are broadly defined and do not necessarily imply a single shared cause. Tightly scoped phenotypes will be best.
Genotype document
Virtually all biological phenomena are at least somewhat heritable. A genotype document starts with a full genome sequence and follows up with modalities for functional context. While this overlaps strongly with the patient-centered document, here we also consider the possibility of incorporating experimental data from patient derived cells (primary or iPSC) and even twins. Since we start with DNA, it makes sense to order modalities bottom-up.
<Whole exome sequence> [Sequence] Pathogenic variant identified in KCNQ1 gene: c.1022C>T (p.Arg341Trp). Heterozygous mutation associated with Long QT Syndrome Type 1 (LQT1).
<Patient-derived iPSC protocol> Extracted patient's peripheral blood mononuclear cells, used Sendai virus vectors to generate iPSCs. Expanded iPSCs in feeder-free conditions using Essential 8 medium. Induced mesoderm formation using CHIR99021 for 24 hours. Cardiac specification using IWR-1 for 48 hours. Cardiomyocyte maturation in RPMI/B27 medium for 10 days. Selection of cardiomyocytes using lactate-supplemented medium for 5 days. Maintained cardiomyocytes in RPMI/B27 medium.
<Compound treatment protocol> Compound: Propranolol (Beta-blocker). Prepared stock solution of propranolol at 10 mM in sterile water. Diluted stock solution to working concentration: 10 µM in cardiomyocyte maintenance medium. Treatment groups: Control (vehicle only), 10 µM propranolol. Added treatment to mature iPSC-derived cardiomyocytes (day 30 post-differentiation). Incubated cells with compound for 48 hours.
<Functional readout> [Time series] Patch clamp readings measuring current passing through cell membranes. Control (untreated) cells: Delayed rectifier potassium current (IKs) reduced compared to healthy controls, prolonged action potential duration (APD), increased propensity for early afterdepolarizations (EADs). Propranolol-treated cells (10 µM, 48 hours): Modest increase in IKs current density, slight shortening of APD, reduced occurrence of EADs, shift in voltage-dependent activation towards more negative potentials.
<Patient health history> 28-year-old female, Caucasian, Height: 165 cm, Weight: 60 kg. Non-smoker, occasional alcohol consumption, regular exercise routine (3-4 times/week, moderate intensity). Diagnosed with Long QT Syndrome at age 16 following a syncopal episode. Current medication: Nadolol 40mg daily. Family history of sudden cardiac death (maternal uncle, age 45). No other significant medical conditions.
<Identical twin health history> 28-year-old female, Caucasian, Height: 164 cm, Weight: 58 kg. Non-smoker, no alcohol consumption, sedentary lifestyle. Asymptomatic, no history of syncope or cardiac events. Currently unmedicated, under regular cardiac monitoring. No other significant medical conditions.
Full genetic sequences are long and haven’t been successfully modeled as a whole. On the other hand, chopping up the DNA sequence may eliminate important loci. Reduced representations like the polygenic risk score vectors used by Med-Gemini might be used instead, although they cannot be used to discover novel associations. Also, notice the inclusion of experimental methods for in vitro experiments, which, as discussed, may alleviate the confounding effect of measurement noise.
Cell type document
A cell type is a grouping of cells with similar biochemical profiles and functional roles in the body. Because the acquisition of many single cell technologies is mutually exclusive, a cell type document is a natural place to put together omics readouts that share a common background if not the exact same population of cells.
<Description> Helper T cells, also known as CD4+ T cells, are a crucial component of the adaptive immune system. They are primarily found in lymphoid tissues such as lymph nodes, spleen, and tonsils, as well as circulating in the bloodstream. Helper T cells play a central role in orchestrating immune responses by: Activating and directing other immune cells, including B cells and cytotoxic T cells, producing cytokines to regulate immune responses, and providing immunological memory for faster responses to previously encountered pathogens. Helper T cells are associated with various diseases, including: HIV/AIDS, where the virus targets and depletes these cells, autoimmune disorders like multiple sclerosis, rheumatoid arthritis, and lupus, and severe combined immunodeficiency (SCID), where these cells fail to develop properly.
<Single cell paired RNAseq and ATACseq> [Interleaved sequence of profiles] RNAseq profiles show: High expression of CD4, IL2RA, FOXP3, moderate expression of CTLA4, TNFRSF18, IL7R, low expression of CD8A, GZMA, PRF1. ATACseq profiles show: Open chromatin regions near CD4, IL2RA, FOXP3 gene loci, partially accessible regions near cytokine genes (IL2, IFNG, IL4), closed chromatin near CD8A and cytotoxic genes.
<Microscopy image 1> [Image] Brightfield microscopy of a resting helper T cell: Small, round cell with a large nucleus and thin rim of cytoplasm, diameter approximately 7-10 μm, smooth cell surface.
<Microscopy image 2> [Image] Fluorescence microscopy of an activated helper T cell, CD4 receptor labeled with green fluorescent antibody, cell surface shows irregular projections (pseudopodia), increased cell size (10-12 μm) compared to resting state.
<Microscopy image 3> [Image] Confocal microscopy of a helper T cell interacting with an antigen-presenting cell. Helper T cell (labeled red) forming an immunological synapse with a dendritic cell (labeled blue). Concentration of CD3 and CD28 receptors (labeled green) at the synapse interface.
<Microscopy image 4> [Image] Electron micrograph of a helper T cell. Detailed view of cellular ultrastructure. Dense, centrally located nucleus with peripheral heterochromatin, scattered mitochondria and rough endoplasmic reticulum in the cytoplasm, presence of secretory vesicles near the cell membrane in an activated cell.
Cell types have subtypes and distinct states. In this example, we emphasize views of Helper T cells at rest and under activation as well as distributions of omics profiles for multiple cells, to hopefully cover some of their functional range. Since all modalities are meant to be at the single cell level, hierarchical ordering wouldn’t quite make sense. For paired omics modalities, we interleave the profiles. The microscopy images are roughly ordered by the level of detail they provide, with the assumption that low-resolution information about the cell type will make the prediction of high-resolution information easier.
Gene document
The gene document combines DNA, RNA, and proteins. Biomolecular sequences and structures as well as known interactions and functional roles (in specific cells or more generally) are incorporated for richer context:
<Description> MTOR (Mechanistic Target of Rapamycin Kinase) is a serine/threonine protein kinase that regulates cell growth, proliferation, motility, and survival. It's a key regulator of cellular metabolism and plays a crucial role in nutrient sensing and response to cellular stress. MTOR is part of two distinct protein complexes: mTORC1 and mTORC2, each with different functions and downstream targets.
<DNA sequence> [Nucleotide sequence] The MTOR gene is located on chromosome 1 (1p36.22) and spans approximately 150 kb. Promoter region: TATAAA...GCGCGC...CAAT… Exon 1: ATGGCTTCGAAGCCATGGTG… Intron 1: GTAAGT...CAGGTG. (A full representation would include all 58 exons and their intervening introns)
<mRNA sequences> [Nucleotide sequences] Primary transcript: 5'-GGAAGAUG...-3'. Alternative splice variant: 5'-GGAC[EXON SKIP]AA..-3'.
<Protein sequence> [Amino acid sequence] MAFEAMV…
<Protein structure> [3D coordinates] MTOR is a large protein (2,549 amino acids) with multiple domains, including: HEAT repeats, FAT domain, FRB domain (rapamycin-binding), Kinase domain, FATC domain.
<Targeting therapeutics> Rapamycin (sirolimus) and rapalogs (e.g., everolimus, temsirolimus): Allosteric inhibitors that bind to FKBP12 and then to the FRB domain of MTOR. ATP-competitive inhibitors (e.g., Torin1, PP242): Direct inhibitors of MTOR's kinase activity. Dual PI3K/MTOR inhibitors (e.g., NVP-BEZ235): Target both MTOR and PI3K pathways. RapaLink-1: Bivalent mTORC1 inhibitor combining features of rapamycin and ATP-competitive inhibitors.
<Pathways and interactions> Insulin/IGF-1 signaling pathway: Activates MTOR through PI3K and AKT. AMPK pathway: Inhibits MTOR activity in response to low energy states. Amino acid sensing: Activates MTOR through Rag GTPases. Hypoxia response: HIF-1α regulates MTOR activity. Downstream effectors: 4E-BP1 and S6K1 (protein synthesis), ULK1 (autophagy regulation), SREBP (lipid synthesis).
<Disease associations> Cancer: Hyperactivation of MTOR is implicated in various cancers, including breast, prostate, and renal cell carcinoma. Metabolic disorders: MTOR dysregulation is associated with obesity and type 2 diabetes. Neurodegenerative diseases: Altered MTOR signaling is observed in Alzheimer's and Parkinson's diseases. Immunological disorders: MTOR plays a role in T cell differentiation and function, impacting autoimmune diseases. Aging: MTOR inhibition has been linked to increased lifespan in model organisms.
Disease associations here link back to disease documents and provide further grounding of potential causes.
Therapeutic document
Therapeutics are commonly tested in cell lines (e.g., phenotypic screens), animal models, and humans. The ability to perform the same intervention and measure outcomes at multiple levels in the biological hierarchy makes this a powerful document type for building translational intuition. Effectively, the document may look a bit like an abbreviated clinical trial report with bottom-up ordering:
<Therapeutic representation> CC(=O)NCCCCCC(=O)NC(CS)C(=O)NC1CCCCC1
<Description> Vorinostat (also known as SAHA) is a potent histone deacetylase (HDAC) inhibitor. It functions by binding to the catalytic domain of HDACs, particularly HDAC1, HDAC2, HDAC3, and HDAC6. This binding inhibits the deacetylation of histones, leading to increased acetylation of histone proteins. The resulting hyperacetylation promotes the expression of genes involved in cell cycle arrest, differentiation, and apoptosis in cancer cells.
<Description of the cell line and preparation protocol> The A549 cell line is a human lung adenocarcinoma cell line derived from the explanted tumor of a 58-year-old Caucasian male. These cells are widely used as an in vitro model for Type II pulmonary epithelial cells and for studying lung cancer. Treatment: Vorinostat was dissolved in DMSO to create a 10 mM stock solution. Working solutions were prepared by diluting the stock in growth medium. Cells were treated with 100 nM Vorinostat or vehicle control (DMSO) for 24 hours.
<Control in vitro> [Cellpaint images] Fluorescence microscopy images of untreated A549 cells show typical morphology of lung adenocarcinoma cells. The nucleus (stained blue with DAPI) appears large and prominent. The cytoplasm (stained green with FITC-conjugated antibodies) shows a spread-out morphology with visible cell boundaries. Mitochondria (stained red with MitoTracker) appear as small, elongated structures dispersed throughout the cytoplasm. The overall cell population shows high density and rapid proliferation characteristic of cancer cells.
<Treated in vitro> [Cellpaint images] After treatment with Vorinostat, A549 cells exhibit significant morphological changes. The nuclei (blue) appear larger and more diffuse, indicating chromatin relaxation due to histone hyperacetylation. The cytoplasm (green) shows signs of shrinkage and membrane blebbing, suggestive of early apoptotic events. Mitochondria (red) appear more fragmented and clustered, potentially indicating mitochondrial dysfunction. The cell population density is noticeably reduced compared to the untreated sample, suggesting decreased proliferation or increased cell death.
<Comparison> Untreated cells display characteristics of rapidly proliferating cancer cells, with compact nuclei and well-defined cytoplasmic structures. In contrast, Vorinostat-treated cells show clear signs of anti-cancer effects. The nuclear enlargement and diffusion observed in treated cells are consistent with the drug's mechanism of action as an HDAC inhibitor, leading to chromatin relaxation. The cytoplasmic changes, including cell shrinkage and membrane blebbing, strongly suggest the induction of apoptosis. The fragmentation and clustering of mitochondria in treated cells may indicate disruption of cellular energy metabolism. Overall, these changes point to Vorinostat's efficacy in inhibiting cell proliferation and promoting cell death in A549 lung cancer cells.
<Pre-treatment population trial> Prior to Vorinostat treatment, patients with advanced Cutaneous T-cell lymphoma (CTCL) who had failed at least two prior systemic therapies were assessed for their baseline skin lesion severity. The severity was measured using the modified Severity Weighted Assessment Tool (mSWAT), which evaluates the type of lesion (patch, plaque, or tumor) and the percentage of body surface area affected. At baseline, the median mSWAT score was 45.5 (range: 10-95), indicating moderate to severe skin involvement. 60% of patients had stage IIB disease or higher.
<Post-treatment population trial> Following a 16-week course of Vorinostat treatment, patients were re-evaluated using the mSWAT. The primary endpoint was the reduction in skin lesion severity, defined as a ≥50% reduction in the mSWAT score from baseline. After treatment, 29.7% of patients achieved a ≥50% reduction in their mSWAT score. The median time to response was 8 weeks (range: 4-14 weeks). Additionally, 32.8% of patients experienced pruritus relief, a key symptom of CTCL.
<Trial result description> The clinical trial results demonstrate a significant improvement in skin lesion severity with Vorinostat treatment in advanced CTCL patients. The observed response rate of 29.7% represents a clear clinical benefit, especially considering these patients had previously failed at least two systemic therapies. This reduction in mSWAT score indicates that Vorinostat has substantial activity against the cutaneous manifestations of CTCL. The relatively rapid median time to response (8 weeks) suggests that patients may experience improvement in a clinically relevant timeframe.d
There is substantial noise in any attempt to translate from findings in cells to animals. It’s likely that adding many studies from the lower levels of the hierarchy will make clinical results easier to predict.
You might be interested in talking to these people: https://www.lesswrong.com/posts/SsLkxCxmkbBudLHQr/tabula-bio-towards-a-future-free-of-disease-and-looking-for